簡介
龐貝氏症(Pompe disease),又名肝醣儲積症第二型(Glycogen Storage Disease, type II),是一種體染色體隱性遺傳罕見疾病,發病率約為五萬分之一至四萬分之一,最早可追溯至西元1932年,荷蘭醫師J.C. Pompe是首位敘述此項疾病的醫師。此種疾病係由於患者的第17號體染色體上的GAA基因變異,無法正常生成酸性α葡萄糖苷酶(acid α glucosidase, GAA)所造成。GAA是一種溶小體酵素,負責在溶小體內分解體內多餘的肝醣。缺乏此種酵素的患者,會使累積在溶小體中的肝醣無法被分解,影響細胞功能,使得肌肉功能受損,進而影響全身肌肉組織及其他器官,最常見的症狀為肌肉無力及心臟肥大等。
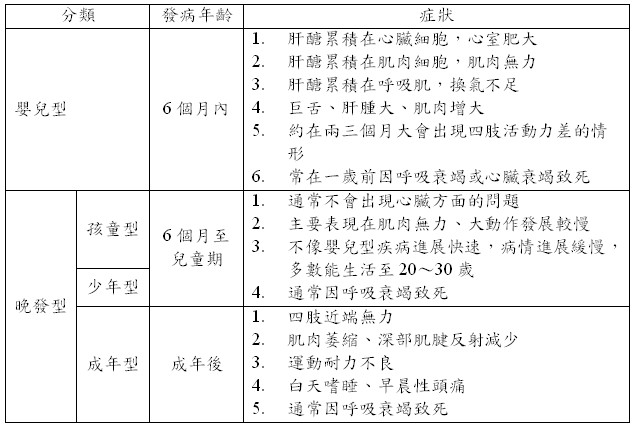
雖然嬰兒一出生就帶有該疾病,但根據發病年齡與對器官的影響,可分為嬰兒型及晚發型,其中晚發型又可細分為孩童型、少年型及成年型,用以下表格來簡單介紹。
龐貝氏症的治療又可分為藥物治療及非藥物治療。龐貝氏症的病因為缺乏GAA酵素,所以常見的藥物治療就是定期施打酵素,也就是Myozyme(Alglucosidase alfa),非藥物治療方面有飲食照顧、呼吸照護、復健治療等。
藥物治療
由於龐貝氏症為缺乏GAA酵素引起,可以藉由定期施打由基因工程製造的藥物Myozyme,來改善病人的症狀。此藥物已在2005年列為公告罕見疾病藥物並獲得健保給付。且歐盟醫藥品管理局(EMEA)以及美國食品藥物管理局(FDA)也於2006年核准此藥上市。
Myozyme(Alglucosidase alfa)
- 適應症:GAA酵素,用於治療GAA酵素缺乏之龐貝氏症患者。
- 標準用量:20mg/kg,每兩個禮拜靜脈注射一次。
- 副作用:蕁麻疹、皮膚紅疹、嘔吐、腹瀉、心搏過速、血壓異常、咳嗽、呼吸急促、貧血、IgG抗體增加、過敏反應等。
- 注意事項:輸注時須注意輸注相關反應,通常會在輸注時或結束後2小時內發生,會有呼及急促、面潮紅、血壓上升、血氧降低等情況發生。當病人出現超敏或過敏反應應立即停止輸注,輕度至中度反應可透過減緩輸注速率,或是使用抗組織胺及類固醇減輕症狀。
飲食照顧
龐貝氏症的患者,常因肌肉無力而造成餵食困難,所以有建議採取高蛋白、低碳水化合物的飲食方法,來改善肌肉功能,但也非對所有的患者都有作用。隨著病情的進展,部分患者會因肌肉無力,導致腸胃功能蠕動變差,嚴重可能導致胃食道逆流而引起吸入性肺炎,此時就需輔佐外在的餵食管,除了使病患能夠充分攝取營養,也能降低併發症的發生。
呼吸照護
由於呼吸肌無力,患者常有呼吸困難的問題,呼吸照護在患者身上是個重要的課題。患者時常無力將痰咳出,在呼吸肌還有力氣時,患者本身也可以藉由胸腔物理治療,進行呼吸訓練,利用腹部的力量把痰咳出來。倘若呼吸肌無力,此時就需要照顧者學習如何排痰、抽痰,維持肺部清潔。
隨著病情的進展或是緊急的狀況發生,必要時須使用呼吸輔助儀器來輔助病人,減輕病人呼吸肌的負擔。呼吸器是藉由幫浦,以不同壓力運送溫暖潮濕的空氣進入肺部。呼吸器的種類繁多,可視患者需求做合適的選擇。
復健治療
由於患者肌肉無力,肌肉會因缺乏運動而僵硬攣縮。此外接受酵素治療的患者,肌肉活動可以幫助酵素進入細胞,所以為患者量身打造運動計畫很重要,包括動作與肌力訓練、被動的關節按摩,必要時可以使用輔助性器材來改善生活品質。
小結
龐貝氏症是一種遺傳性疾病,可透過基因檢測發現。目前臺大醫院新生兒篩檢中心有提供此項服務,且有研究指出早期治療對患者的存活有大幅的改善。另外除了上述的治療之外,別忘了注意患者的心理健康,由於肌肉無力造成行動上的不便,患者更需要大家的關心與支持。目前社會上也有提供許多的相關資源可以給病患及家屬參考,例如財團法人罕見疾病基金會、社團法人龐貝氏症協會、國際龐貝氏協會等。
參考資料
- 財團法人罕見疾病基金會(2006)‧龐貝氏症照顧手冊‧台北縣新莊市:衛生署國民健康局。
- http://www.genes-at-taiwan.com.tw/genehelp/database/Disease/Pompe_new7_20110302.htm (2016年12月25日)
藥劑部藥師 陳翠容
專業諮詢:基因醫學部主治醫師 簡穎秀 |
|

