根據衛生福利部統計,癌症為國人十大死因的第一位,民國104年癌症(惡性腫瘤)死亡人數為46,829人,占所有死亡人數的28.6%,十大癌症死亡率依序為:(1)氣管、支氣管和肺癌;(2)肝和肝內膽管癌;(3)結腸、直腸和肛門癌;(4)女性乳房癌;(5)口腔癌;(6)前列腺(攝護腺)癌;(7)胃癌;(8)胰臟癌;(9)食道癌;(10)子宮頸及部位未明示子宮癌。因此,癌症的防治在現代人,顯得相當重要。當家族中有成員得到癌症,有許多人不禁會問,我是不是也有癌症的風險?或是這個癌症會不會遺傳?所以我們今天要探討這個主題。
何謂遺傳性癌症?
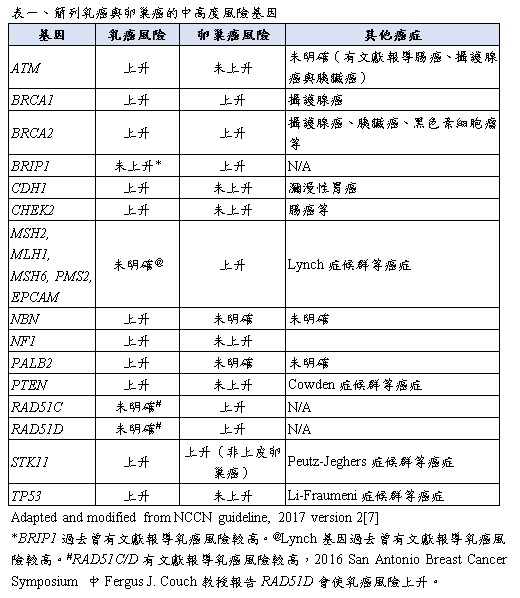
遺傳性癌症在所有癌症病患中,其實僅占一小部分族群,但仍不能忽視。根據過去一項大規模研究,在33,197位癌症病患中,有14.6%的病患有家族癌症的病史,而有7.7%的病人,具有強烈的家族癌症病史[1],也就是說,大約一成左右的癌症病患,屬於家族性癌症或是遺傳性癌症[1, 2]。根據美國臨床腫瘤醫學會,針對遺傳性癌症(hereditary cancer)的定義表示,是由於單基因發生致病性突變,導致有很高的罹癌風險(defined here as having a risk almost entirely attributable to germline mutations in a single gene)。遺傳性癌症與家族性癌症的定義界限並不十分清楚,一般而言,常見被提及有遺傳性的癌症例如乳癌,約有5-10%的病患屬於遺傳性癌症[3],另有約15%的病患,屬於家族性的癌症,並非由目前已知的單基因突變所引起,可能是由於多基因遺傳與後天環境因子交互作用而形成。因此,所謂的癌症易感基因(cancer susceptibility gene或稱為predisposing gene),可以依照癌症的外顯率(penetrance)分成高風險(high penetrance)、中度風險(moderate penetrance)與低風險(low penetrance)基因[3-5]。高風險基因,例如TP53、BRCA1、BRCA2與PTEN等基因突變,所造成高度罹癌的風險;中度風險,例如RAD51C、CHEK2等基因的突變;而低風險基因變異,大多是過去genome-wide association study (GWAS)所找到的Single Nucleotide Polymorphism(SNP)。目前依照美國臨床腫瘤醫學會與NCCN guideline的建議,只有moderate-to-high penetrance的這些(中度到高度風險)基因,被認為在臨床上需要檢驗,也只有這些基因突變的帶因者,因其罹癌風險顯著上升,需要較早與較頻繁的頻率,做標的器官的癌症篩檢或是其他預防措施;方式則因不同基因而不同[6, 7]。
遺傳性癌症病人的臨床表現,常具有一些特徵,利用這些特徵,可以協助醫師與病人本身,發現是否可能屬於遺傳性癌症的病人。一般而言,可以區分為病人個人特徵與家族特徵兩個方面,病人方面主要可以歸類成四種[8]:(1)多發腫瘤,可以是同一器官有多個原發腫瘤(轉移的不算),或是成對的器官有兩個以上原發腫瘤,例如雙側乳房都長出乳癌(時間上可以是先後發生);另一種情形是同一人,有兩種以上的癌症(時間上可以是先後發生),例如同一人有乳癌、卵巢癌、大腸癌等兩種以上癌症。(2)早發腫瘤,指的是癌症發生的年齡,明顯早於該種癌症一般發生的年齡,例如小於35歲得到乳癌。(3)罕見腫瘤,包含了癌症發生在不常見之性別上,常提及的即是男性發生乳癌;少見的腫瘤組織型態,也意味著可能與特殊基因變異有關,或是某一些內分泌腫瘤,例如胰臟神經內分泌腫瘤。(4)合併先天已知的基因疾病。家族特徵方面,同源血親中,有多人得到相同或是相關的癌症。因此,在遺傳性癌症評估的第一步,即是由遺傳諮詢師與醫師,檢視病人癌症的特徵與繪製家族成員癌症的分布狀況,來釐清是否有遺傳性癌症的可能,此步驟就是遺傳諮詢(Genetic counseling)[9, 10]。
遺傳性癌症門診與處理流程
如圖一與上段文章所述,當有上述特徵的癌症病患與家族成員,來到遺傳性癌症門診時,遺傳諮詢師與醫師會先仔細評估,稱之為遺傳諮詢(Genetic counseling)[9, 10],當懷疑病患是遺傳性癌症時,會先說明可能是哪一症候群或是該特徵符合哪一些癌症症候群,使病患與家屬了解該癌症症候群的罹癌種類與風險,若是有目前已知的基因可以提供檢驗,則可以考慮執行檢驗,至於是否要接受基因檢驗,由病人與其家族成員自行決定。基因檢驗為個人之隱私,待結果出爐,原則上須由病患本人親自領取;基因檢驗後,有後續的post-genetic counseling,包含結果的解說。由於基因檢驗結果可能是陽性、陰性或是無法判定,因此須由專業的醫師與遺傳諮詢師解說。當病人確定帶有該癌症易感基因突變時,門診將給予心理支持、與後續的預防措施,包含有預防性的治療或是給予具有實證醫學或是治療準則建議的器官癌症篩檢,來降低罹癌相關的死亡率。

常見的遺傳性癌症症候群
常見的遺傳性癌症症候群有遺傳性乳癌與卵巢癌、遺傳性大腸癌等。本文舉上述兩個例子做基本介紹。
1. 遺傳性乳癌與卵巢癌
遺傳性乳癌與卵巢癌,是最為人所知的例子,好萊塢知名女星安潔莉娜‧裘莉,因為母親與阿姨分別因卵巢癌與乳癌過世,接受基因檢測;並得知自己有BRCA1基因突變,已於2013年初接受預防性雙乳房切除重建手術,接著接受預防性雙側輸卵管及卵巢切除手術。臨床上,根據最新NCCN guideline的建議[7],當一位乳癌病患,具有下述特徵時,建議接受遺傳諮詢是否有遺傳性乳癌與卵巢癌的可能性:(1)已知帶有基因變異者;(2)個人或是家族有卵巢癌者;(3)個人或是家族有雙側乳癌者;(4)早發性乳癌;(NCCN指的是小於或是等於40歲);(5)三陰性乳癌且年紀小於60歲;(6)家族史中,有另一位血親具有小於50歲發病的乳癌,或是至少有另外兩位乳癌患者(不限年紀);(7)家族史中有胰臟癌者;(8)家族史中有男性乳癌;(9)非上述癌症,家族中(含病人本身)有三位癌症患者或是三種癌症的患者,例如腸胃道癌症、子宮內膜癌、男性攝護腺癌、血癌等等;以上皆被建議接受癌症遺傳諮詢。
致病機轉
促成遺傳性乳癌與卵巢癌症候群(Hereditary breast and ovary cancer syndrome)最重要的兩個基因是BRCA1與BRCA2,BRCA1與BRCA2是1990年代在美國發現的[11, 12]。1990年,Hall等人研究早發與遺傳性乳癌家族,經由連鎖遺傳學定律(linkage analysis),發現染色體17q21與早發家族性乳癌有高度相關。其後1994年,Miki等人證實染色體17q21上的BRCA1基因為造成乳癌(與卵巢癌)的基因。同年,Wooster等人發現位於13q12 -13也與乳癌發生相關,因而找到BRCA2基因[13]。BRCA1有24個exon,轉譯而成的BRCA1蛋白質共有1863個胺基酸[13];BRCA2有27個exon,轉譯而成的BRCA2蛋白質共有3418個胺基酸[13]。這兩個基因是屬於抑癌基因(tumor-suppressor gene),負責雙股DNA損壞的修復機轉。當細胞內雙股DNA損壞,細胞有兩個方式負責修補,第一個修復的方式稱為同源重組(Homologous Recombination)[11],另一個是非同源染色體結合(non-Homologous end-joint),只有經由同源重組的方式來修復,雙股DNA才可以正確無誤的修復。而BRCA1與BRCA2所參與的雙股DNA修復機轉,即是同源重組,因此,若這兩個基因其中之一發生缺陷,則雙股DNA受到攻擊斷裂後,會無法正確修復。當細胞內DNA壞損累積到一定程度,細胞就會發生癌變。同源重組的修復(Homologous Recombination repair),有許多蛋白質參與其中,主要為Fanconi’s pathway相關蛋白質[11],近年來研究得知,參與同源重組的基因發生致病性變異[14],也會發生類似BRCA1與BRCA2突變的表現型,也就是與乳癌、卵巢癌或是相關癌症的發生。
多基因篩檢
由上可知,參與遺傳性乳癌的基因,不僅是BRCA1與BRCA2;除了上述同源重組基因變異所導致之外,還有許多其他症候群也會導致遺傳性乳癌,例如遺傳性瀰漫性胃癌(Hereditary diffuse gastric cancer),導因於CDH1基因變異,該疾病亦會發生遺傳性乳癌[15]。Cowden症候群,導因於PTEN基因變異,亦可造成遺傳性乳癌,且該兩症候群基因變異時,對乳癌的發生皆是屬於高外顯率(high penetrance)[16, 17]。另外,遺傳性大腸癌中的Lynch症候群,卵巢癌的風險也會顯著上升[18, 19]。近年來基因檢驗技術也大幅進步,隨著次世代定序技術(next-generation sequencing)的使用,目前對於遺傳性癌症症候群的基因檢驗,可以同步進行多基因的篩檢,在NCCN guideline中,也被建議考慮使用。臺大醫院從兩年前,即已經建立遺傳性癌症的多基因檢驗[20],對於遺傳性乳癌與卵巢癌,主要檢驗的基因有ATM、BRCA1、BRCA2、BRIP1、CHD1、CHEK2、NBN、PALB2、PTEN、RAD51C、RAD51D、STK11、TP53與Lynch症候群基因(MSH2, MLH1, MSH6, PMS2, EPCAM)等已經在NCCN guideline提到防治的基因。其他例如BARD1、FANCC、MRE11A、RECQL、RAD50、SLX4、SMARCA4或XRCC2等在文獻中也提到造成遺傳性乳癌或是卵巢癌者,也一併檢驗,目前可以檢驗的遺傳性癌症(不限乳癌)基因總數約80個,持續增加中。唯目前基因檢驗並非健保給付項目,需自費。
預防與治療
對於預防晚期癌症的發生與降低癌症相關的死亡率,對於這些基因變異的帶因者,過去有諸多的研究。預防措施一般可以分成三種:(1)標的器官的檢查(target organ screening);(2)標的器官的預防性切除(prophylactic surgery);(3)預防性投藥(chemoprevention)[7]。
對於BRCA1與BRCA2基因變異的帶因者,NCCN guideline建議[7]:(1)女性應從25歲開始接受乳房的篩檢,25到29歲期間,考慮每年核磁共振造影檢查;(2)30歲之後(至75歲),應考慮每年接受乳房攝影與核磁共振造影檢查,在臺灣目前的臨床實務,乳房超音波檢查,也是可以考慮的選擇;(3)若是一側乳房已經得到乳癌,在該側乳癌接受切除與後續的輔助治療(例如化學治療)之後,對側乳房也應持續影像學追蹤,過去的研究顯示,帶因者接受對側乳房預防性切除,長期追蹤的結果,證實可以降低乳癌相關的死亡率;(4)預防性雙側乳房切除與重建手術,是過去研究證實可以降低乳癌相關死亡率的一項重要措施,但是該預防措施,需與帶因者詳細溝通,帶因者充分了解,心理諮商後,才予以進行;(5)藥物預防方面,NSABP P-1 trial (National Surgical Adjuvant Breast and Bowel Project Breast Cancer Prevention trial )研究顯示,Tamoxifen可以降低BRCA2突變帶因者發生乳癌的風險[21];(6)卵巢癌方面,NCCN guideline建議,可以待生育下一代結束之後,35-40歲左右,考慮卵巢與輸卵管預防性切除;替代的方法,為規律性的經陰道超音波與腫瘤指數CA-125的檢查,唯這樣的措施並沒有足夠的醫學證據證明可以顯著降低卵巢癌相關的死亡率,而是專家意見的結果;(7)男性帶因者,亦有顯著高於一般男性得到乳癌的風險,故35-40歲之後,應開始接受乳房攝影或是其他影像學檢查。45歲之後,對於BRCA2的突變帶因者,應考慮攝護腺癌的篩檢。
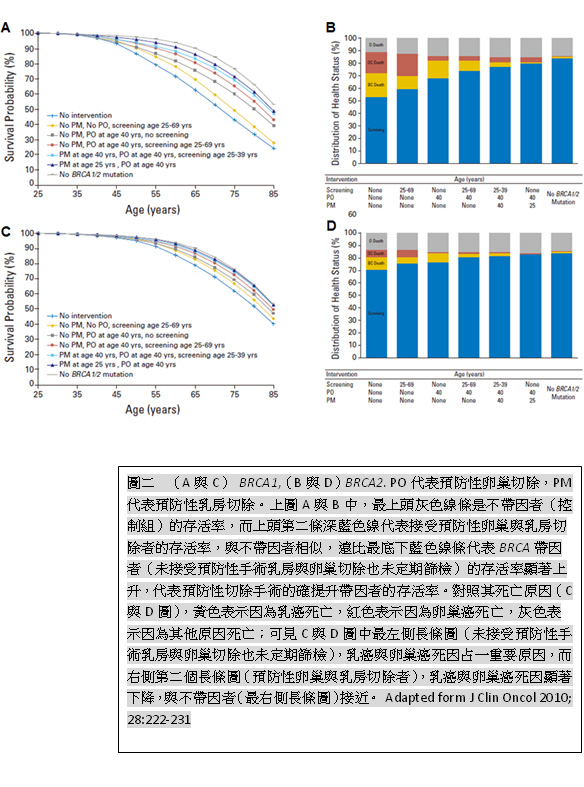
帶因者並不需要特別恐慌,只要接受上述的建議,可以有效的降低癌症相關的死亡率[22],可以由圖二的大規模研究證實,接受規律篩檢或是預防性手術者的存活率,幾乎與一般人相同。其他基因方面,造成乳癌、卵巢癌或是其他癌症罹病上升的風險,整理如表一。
近年來,BRCA一直是熱門的研究題材,由於BRCA基因突變的腫瘤細胞,其基因體不穩定度比較高(genomic instability)[23],雙股DNA修復機轉有缺陷,可以成為治療時的參考。在BRCA相關的癌症治療方面,轉移性乳癌在英國TNT第三期人體臨床試驗,比較使用鉑金與紫杉醇做為第一線化學治療,試驗結果分別在2014與2016年的San Antonio Breast Cancer Symposium報告,發現鉑金治療有顯著較佳的反應率,延緩疾病惡化的存活率與總體存活率。Fong PC等人提出了Poly(ADP-Ribose) Polymerase (PARP)抑制劑(inhibitor),其原理是抑制DNA的單股修復機轉(PARP inhibitor能抑制DNA的單股修復)[24];當DNA的單股與雙股(因BRCA突變)修復機制都失效時,癌症細胞會因為DNA損壞(例如在化學治療之下)而死亡[25]。2014年底,美國FDA已經核准第一個PARP inhibitor (Olaparib)用於BRCA基因突變的卵巢癌[26]。在數個臨床試驗都顯示,使用該藥物,有助於卵巢癌的控制與延長病人存活率[27, 28]。PARP inhibitor對於BRCA基因突變的乳癌,目前則有多個第二、三期人體臨床試驗在進行[29-31],其他BRCA基因突變相關的癌症(例如攝護腺癌),亦有臨床試驗進行中[32]。
2. 遺傳性大腸癌
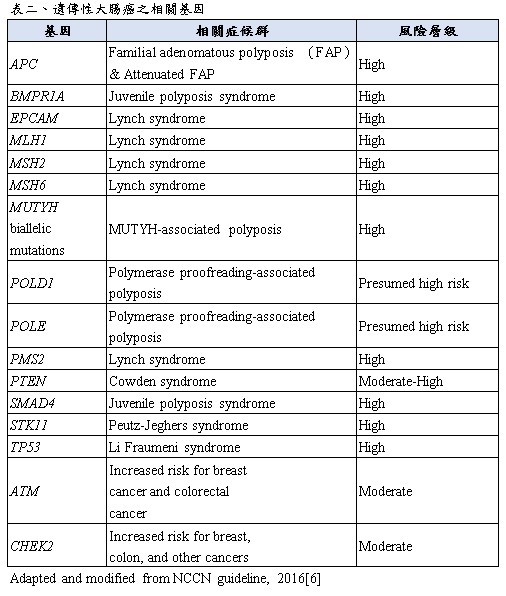
遺傳性大腸癌並非是單一疾病,而是多種症候群與多種已知基因突變皆會造成遺傳性大腸癌。因篇幅關係,簡述並整理如表二。可以依照是否有大腸瘜肉,約略分成兩大類症候。具有腸瘜肉的有Familial adenomatous polyposis (家族遺傳性大腸瘜肉症候群,FAP)、MUTYH-associated polyposis、Polymerase proofreading-associated polyposis、Juvenile polyposis syndrome與Peutz-Jeghers syndrome等症候群。非瘜肉型的有Lynch syndrome、Li Fraumeni syndrome、Cowden syndrome、ATM與CHEK2等基因突變等[6]。
家族遺傳性大腸瘜肉症,導因於APC基因發生突變[33],為自體顯性遺傳,當病患身上超過100個以上的瘜肉時,便可以診斷為該疾病;患者在平均年齡十幾歲的時候會開始發生瘜肉。超過 95%的患者在35歲時,就已經有多個腸瘜肉發生。如果FAP病患沒有及早治療,幾乎100%會發展成大腸癌。FAP有幾種臨床表現的變異型,如Attenuated FAP (AFAP)是較溫和的形式,具有AFAP會有較少的大腸瘜肉,但也會增加得到大腸癌的風險;Gardner's Syndrome為典型的FAP,患者會有骨肉癌或軟組織腫瘤;另一變異型為Turcot Syndrome,與某些腦部腫瘤相關。MUTYH基因若是雙股皆發生突變(bi-allelic mutation),則帶因者亦會產生為數眾多的大腸瘜肉,並有高度的大腸癌風險,若是僅單股突變(heterozygous mutation),依照過去的研究,則偏向中低度的大腸癌風險[34, 35]。Peutz-Jeghers syndrome為STK11基因突變[36],其臨床特徵為腸胃道瘜肉及皮膚黏膜的黑色素沉著與腸胃道缺陷(hamartoma),突變帶因者常可見嘴唇有黑色素細胞沉積,該症候群與乳癌、胃腸道惡性腫瘤等癌症相關。
Lynch症候群又稱為遺傳性非瘜肉結直腸癌(HNPCC,Hereditary non-polyposis colorectal cancer),約占大腸直腸癌的3%左右。該症候群是因為DNA修復機轉mismatch repair(MMR)相關基因發生缺陷所導致,目前已知明確會引起該症候群的基因有MSH2、MLH1、MSH6、PMS2與EPCAM[6, 37]。EPCAM基因則是當其在細胞內缺時,造成MSH2基因的啟動子(promoter)被高度甲基化(hypermethylation),造成MSH2基因不活化而導致Lynch症候群。細胞內MMR失去功能將導致基因複製時鹼基之誤配無法修補,造成基因突變,促成癌症;該腫瘤有一特性,即因MMR缺損,導致DNA微小衛星不穩定(MSI,microsatellite instability)。Lynch症候群病患的大腸直腸癌、子宮內膜癌和卵巢癌的風險較一般人顯著上升。臨床上要懷疑一位大腸直腸癌的病人屬於Lynch症候群,可以由家族史及腫瘤特徵來著手[38]。過去由1991年訂定的阿姆斯特丹準則來預測Lynch症候群,1997年有Bethesda準則,目前改版的Bethesda準則建議(1)小於50歲的大腸直腸癌,(2)同時或不同時發生2個以上之大腸癌或Lynch症候群相關癌症,(3)小於60歲的大腸直腸癌且癌組織有微小衛星不穩定型病理變化(MSI-High),(4)大腸直腸癌患者,一等親有一位以上在50歲前有大腸癌或Lynch症候群相關癌症,(5)大腸直腸癌患者,其一等親或二等親至少有2位患有大腸直腸癌或Lynch症候群的相關癌症。另外,當一位大腸直腸癌患者,腫瘤檢測發現高度微小衛星不穩定型病理變化(MSI-H),也建議接受Lynch症候群的篩檢[38]。基因篩檢發現是突變帶因者,依照目前NCCN guideline建議也應及早進行大腸鏡篩檢與子宮卵巢的檢查[6, 37]。
總結
遺傳性癌症雖然是所有癌症病患中的一小族群,但是由於他們具有高罹癌風險,因此不容忽視。可以經由病患的家族或是腫瘤病理特徵,及早發現這一族群,進行基因診斷確認,並應與專科醫師討論,及早進行篩檢或是其他預防措施。如此一來,罹癌而導致的死亡率可以大幅改善,不需再懼怕遺傳性癌症。
參考文獻
- Scheuner MT, McNeel TS, Freedman AN. Population prevalence of familial cancer and common hereditary cancer syndromes. The 2005 California Health Interview Survey. Genet Med 2010; 12: 726-735.
- Schiffman JD. Hereditary cancer syndromes: if you look, you will find them. Pediatr Blood Cancer 2012; 58: 5-6.
- Mavaddat N, Antoniou AC, Easton DF, Garcia-Closas M. Genetic susceptibility to breast cancer. Mol Oncol 2010; 4: 174-191.
- Beggs AD, Hodgson SV. Genomics and breast cancer: the different levels of inherited susceptibility. Eur J Hum Genet 2009; 17: 855-856.
- Cobain EF, Milliron KJ, Merajver SD. Updates on breast cancer genetics: Clinical implications of detecting syndromes of inherited increased susceptibility to breast cancer. Semin Oncol 2016; 43: 528-535.
- Provenzale D, Gupta S, Ahnen DJ et al. Genetic/Familial High-Risk Assessment: Colorectal Version 1.2016, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw 2016; 14: 1010-1030.
- Daly MB, Pilarski R, Berry M et al. NCCN Guidelines Insights: Genetic/Familial High-Risk Assessment: Breast and Ovarian, Version 2.2017. J Natl Compr Canc Netw 2017; 15: 9-20.
- Lindor NM, McMaster ML, Lindor CJ et al. Concise handbook of familial cancer susceptibility syndromes - second edition. J Natl Cancer Inst Monogr 2008; 1-93.
- Christinat A, Pagani O. Practical aspects of genetic counseling in breast cancer: lights and shadows. Breast 2013; 22: 375-382.
- Hampel H, Bennett RL, Buchanan A et al. A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: referral indications for cancer predisposition assessment. Genet Med 2015; 17: 70-87.
- Ceccaldi R, Sarangi P, D'Andrea AD. The Fanconi anaemia pathway: new players and new functions. Nat Rev Mol Cell Biol 2016; 17: 337-349.
- Nielsen FC, van Overeem Hansen T, Sorensen CS. Hereditary breast and ovarian cancer: new genes in confined pathways. Nat Rev Cancer 2016; 16: 599-612.
- Venkitaraman AR. Linking the cellular functions of BRCA genes to cancer pathogenesis and treatment. Annu Rev Pathol 2009; 4: 461-487.
- Walsh CS. Two decades beyond BRCA1/2: Homologous recombination, hereditary cancer risk and a target for ovarian cancer therapy. Gynecol Oncol 2015; 137: 343-350.
- van der Post RS, Vogelaar IP, Manders P et al. Accuracy of Hereditary Diffuse Gastric Cancer Testing Criteria and Outcomes in Patients With a Germline Mutation in CDH1. Gastroenterology 2015; 149: 897-906 e819.
- Mester JL, Moore RA, Eng C. PTEN germline mutations in patients initially tested for other hereditary cancer syndromes: would use of risk assessment tools reduce genetic testing? Oncologist 2013; 18: 1083-1090.
- Ngeow J, Liu C, Zhou K et al. Detecting Germline PTEN Mutations Among At-Risk Patients With Cancer: An Age- and Sex-Specific Cost-Effectiveness Analysis. J Clin Oncol 2015; 33: 2537-2544.
- Chui MH, Ryan P, Radigan J et al. The histomorphology of Lynch syndrome-associated ovarian carcinomas: toward a subtype-specific screening strategy. Am J Surg Pathol 2014; 38: 1173-1181.
- Harkness EF, Barrow E, Newton K et al. Lynch syndrome caused by MLH1 mutations is associated with an increased risk of breast cancer: a cohort study. J Med Genet 2015; 52: 553-556.
- Lin PH, Kuo WH, Huang AC et al. Multiple gene sequencing for risk assessment in patients with early-onset or familial breast cancer. Oncotarget 2016; 7: 8310-8320.
- King MC, Wieand S, Hale K et al. Tamoxifen and breast cancer incidence among women with inherited mutations in BRCA1 and BRCA2: National Surgical Adjuvant Breast and Bowel Project (NSABP-P1) Breast Cancer Prevention Trial. JAMA 2001; 286: 2251-2256.
- Kurian AW, Sigal BM, Plevritis SK. Survival analysis of cancer risk reduction strategies for BRCA1/2 mutation carriers. J Clin Oncol 2010; 28: 222-231.
- Watkins JA, Irshad S, Grigoriadis A, Tutt AN. Genomic scars as biomarkers of homologous recombination deficiency and drug response in breast and ovarian cancers. Breast Cancer Res 2014; 16: 211.
- Fong PC, Boss DS, Yap TA et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med 2009; 361: 123-134.
- Iglehart JD, Silver DP. Synthetic lethality--a new direction in cancer-drug development. N Engl J Med 2009; 361: 189-191.
- Kim G, Ison G, McKee AE et al. FDA Approval Summary: Olaparib Monotherapy in Patients with Deleterious Germline BRCA-Mutated Advanced Ovarian Cancer Treated with Three or More Lines of Chemotherapy. Clin Cancer Res 2015; 21: 4257-4261.
- Ledermann JA, Harter P, Gourley C et al. Overall survival in patients with platinum-sensitive recurrent serous ovarian cancer receiving olaparib maintenance monotherapy: an updated analysis from a randomised, placebo-controlled, double-blind, phase 2 trial. Lancet Oncol 2016; 17: 1579-1589.
- Matulonis UA, Penson RT, Domchek SM et al. Olaparib monotherapy in patients with advanced relapsed ovarian cancer and a germline BRCA1/2 mutation: a multistudy analysis of response rates and safety. Ann Oncol 2016; 27: 1013-1019.
- van der Noll R, Marchetti S, Steeghs N et al. Long-term safety and anti-tumour activity of olaparib monotherapy after combination with carboplatin and paclitaxel in patients with advanced breast, ovarian or fallopian tube cancer. Br J Cancer 2015; 113: 396-402.
- Park JW, Liu MC, Yee D et al. Adaptive Randomization of Neratinib in Early Breast Cancer. N Engl J Med 2016; 375: 11-22.
- Rugo HS, Olopade OI, DeMichele A et al. Adaptive Randomization of Veliparib-Carboplatin Treatment in Breast Cancer. N Engl J Med 2016; 375: 23-34.
- Mateo J, Carreira S, Sandhu S et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N Engl J Med 2015; 373: 1697-1708.
- Buecher B. Colorectal adenomatous polyposis syndromes: Genetic determinism, clinical presentation and recommendations for care. Bull Cancer 2016; 103: 199-209.
- Borras E, Taggart MW, Lynch PM, Vilar E. Establishing a diagnostic road map for MUTYH-associated polyposis. Clin Cancer Res 2014; 20: 1061-1063.
- Yamaguchi S, Ogata H, Katsumata D et al. MUTYH-associated colorectal cancer and adenomatous polyposis. Surg Today 2014; 44: 593-600.
- Meserve EE, Nucci MR. Peutz-Jeghers Syndrome: Pathobiology, Pathologic Manifestations, and Suggestions for Recommending Genetic Testing in Pathology Reports. Surg Pathol Clin 2016; 9: 243-268.
- Yamano T, Hamanaka M, Babaya A et al. Management strategies in Lynch syndrome and familial adenomatous polyposis: a national healthcare survey in Japan. Cancer Sci 2016.
- Rubenstein JH, Enns R, Heidelbaugh J et al. American Gastroenterological Association Institute Guideline on the Diagnosis and Management of Lynch Syndrome. Gastroenterology 2015; 149: 777-782; quiz e716-777.
|

